

美国干细胞市场法律法规分析(80页报告)
或扫码获取报告

美国联邦与州政府对干细胞产业的监管态度存在差异,政策曾有反复,但美国在临床试验数量及市场规模上仍保持全球领先。联邦层面,2015年《21世纪治愈法案》、2017年FDA再生医学政策框架(含四份科学监管指导文件)、2022-2023年《国家生物技术和生物制造计划》等政策,旨在减少生物技术依赖并明确发展目标。州层面如得克萨斯州、犹他州允许医疗机构使用未经FDA批准的胎盘干细胞疗法,仅需告知患者“未获批准”并获知情同意。美国已形成较为完善法规框架,由上位法律、法规、管理制度与指南三层组成,将细胞组织产品(HCT/Ps)分为PHS 351(CBER审批)与PHS 361(医院直接临床应用)两类管理,具体监管机构详见表2-1。


法律层面,干细胞治疗管理基于《美国食品、药品和化妆品法案》(FD&CAct)及《公共卫生服务法案》(PHSAct)两部国会法案,《美国联邦条例》(CFR)为FDA管理基础。核心法规包括研究性新药申请规定(21CFR312)及现行药品生产质量管理规范(21CFR210&211),后者原则适用于干细胞产品的设备与生产流程。2016年《21世纪治愈法案》支持精准治疗,要求FDA加速审批,新增“再生医学先进疗法(RMAT)”通道,并赋予已上市药物新增罕见病 适应证6个月市场专营权。
二、法规
(一)联邦立法
法规层面,美国2001年发布《联邦法规》第21篇第1271部分(CFRPart1271),2005年实施,为干细胞治疗审批主要依据。该法规将人体细胞组织分为PHS351与PHS 361两类:PHS 351产品属HCT/Ps分类监管,含骨、韧带、外周血干细胞等;PHS361产品无需IND申请,不含HCT/Ps分类,含全血成分、动物来源细胞及同源性应用骨髓等,具体分类情况见表2-2。

(二)州级立法
美国作为联邦制国家,州级立法与联邦监管存在冲突。截至2025年6月,明确允许特定条件下开展未经FDA批准干细胞疗法的州主要有五个。
1.犹他州(Utah)
法案名称:《胎盘组织修正案》(Placental Tissue Amendments)
生效时间:2024年5月1日
核心内容:允许医疗提供者(医生、自然疗法师等)使用未经FDA批准的胎盘干细胞疗法,仅需声明“未经FDA批准”并获取知情同意,无需安全性或有效性审查。
2.得克萨斯州(Texas)
法案名称:《实验性干细胞治疗法案》(Experimental Stem Cell Therapy Act)
生效时间:2017年9月1日
核心内容:允许严重慢性病或绝症患者在医生推荐及机构审查委员会(IRB)监督下使用成人干细胞(如自体脂肪干细胞),要求疗法需在人类临床试验中测Y试过(地点不限美国)。
3.密西西比州(Mississippi)与北卡罗来纳州(North Carolina)
法案类型:“尝试权法案"(Right-to-Try)扩展
生效时间:2020年后(具体年份未明确)
核心内容:允许绝症患者使用经人体测试(不限于美国试验)的成体干细胞疗法,需医生推荐及IRB监督;与犹他州的区别在于要求提供初步临床证据,而非完全无审查。
化研究院
4.佛罗里达州(Florida)
法案名称:SB1768法案
生效时间:2025年(具体月份未明确)
核心内容:聚焦成体干细胞与脐带血来源,推动再生医学发展,强调“符合本州价值观”,具体细则尚未公开,旨在平衡技术发展与患者权益。
三、指南与规范
FDA与干细胞领域管理部门、企业及研究机构通过沟通形成制造和临床试验的指南规范,界定的原则适用于细胞疗法评估。FDA与NIH通过签署谅解备忘录(MOU)促进干细胞管理建议的形成。
美国FDA为便于监管和指导产品研发,对上述法规和监管内容进行了原则性技术解读,形成了针对不同问题的“指导原则”。在干细胞产品研发方面,FDA已出台20多个技术性指导原则,包括共性内容(1998年人体细胞及基因治疗产品、2008年CMC及一般安全性文件、2011年细胞产品效应试验与GTP规范、2012年临床前研究、2013年早期临床试验设计草案)及特定疾病/问题指导原则(2010年心脏疾病治疗用细胞产品、2014年手术步骤区分及美容用脂肪来源间充质干细胞监管界定),具体详见表2-3
四、审批监管框架
细胞治疗的临床试验审批由生物制品评估中心(CBER)负责,遵循FDA药品管理法规21CFR312.23,与其他生物制品一致。CBER下设细胞、组织与基因治疗办公室,其中细胞与基因治疗部负责产品审批与准入,采用分级分类管理确保安全性和有效性。未获批产品须标注为研究用,禁止宣传,并需完成I、II、I期临床试验。上市需提交生物制品许可证申请(BLA)或新药申请(1NDA),审批通过后方可进入市场。快速审批程序时间为6~10个月,目前美国已有多种
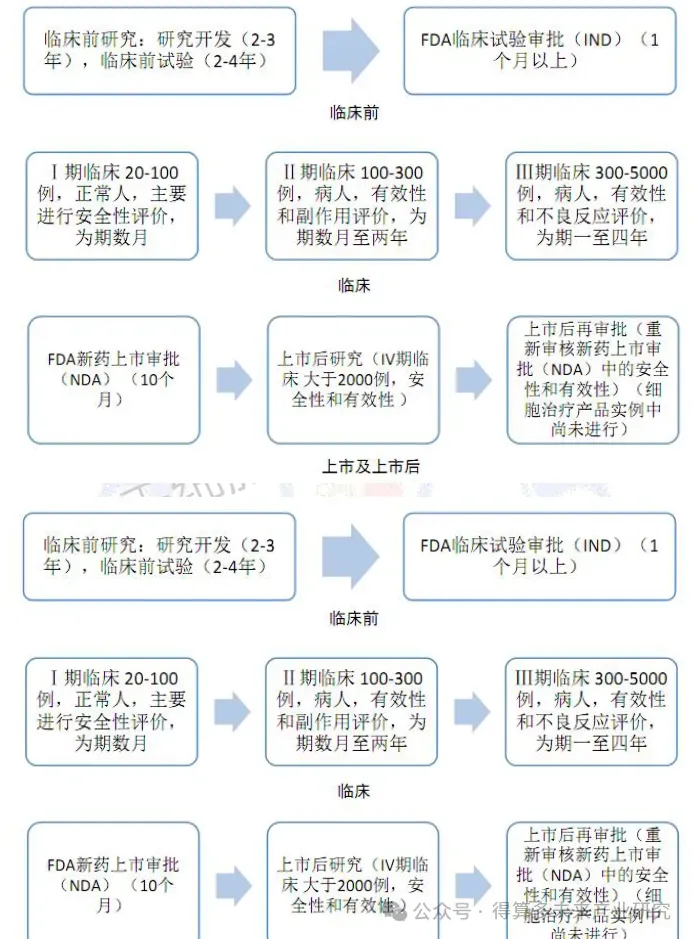
五、特别审批监管2架
美国FDA有五条特别审批通道,即快速通道、加速批准、优先审评、突破性治疗及再生医学先进疗法(RMAT)认定。进入特别通道的条件为:目前无有效药物且新药能填补空白,或新药在有效性/安全性上有明显优势。
目前上市产品主要使用突破性疗法和优先审评途径。优先审评适用于严重疾病及普通疾病,关键在于是否优于现有治疗手段,需药企主动申请,FDA在45天内答复。优先审评仅加速审评阶段(周期6个月),不加速临床试验,标准审评周期为10个月(根据Prescription Drug User Act)。
2012年7月9日,《FDA安全与创新法案》(Food and Drug AdministrationSafetyand Innovation Act)正式实施,FDA第四条特别审批通道诞生,即突破性治疗。突破性治疗资格申请可以与IND一同提交,或在IIND提交后任何阶段,FDA在收到申请60天内给予答复。FDA不会公开申请者名单,也不会公布授予“突破性药物”资格的名单,因为FDA不能公开ID信息。突破性治疗的认定需满足两个条件,即(1) 适应证是严重或致死性疾病;(2)有证据显示在某一重要临床终点上明显优于现有药物。“突破性药物”与“快速通道”几乎模一样,都是为了加速严重或致死性疾病药物的研发与审批,而不同点在于取得认定所需要提供的证据。这里引用FDA的原话:“突破性药物”的认定比“快速通道”更加严格,享有“快速通道”的所有权利,能得到FDA更加密切的指导,当然如果一个药物获得了“突破性药物”,就不需要再申请“快速通道”如果一个药物申请“突破性药物”失败,FDA不会自动启动“快速通道”认定程序,研发单位需要重新申请。
美国药品监督管理局(FDA)于2016年12月13日在21世纪治愈法案(21stCentury Cures Act)中颁布并开始实施的一种研发与审评加速通道,即再生医学先进疗法RMAT认定,旨在加快用于治疗严重疾病的药物的研发与审评,从而尽早满足患者的治疗需求。获得RMAT的药物必须同时满足下列条件:(1)该药物是一种再生医学疗法,定义为细胞疗法或基因治疗、治疗性组织工程产品、人体细胞和组织产品,或使用此类疗法或产品的任何组合产品;(2)该药物旨在治疗、改变、逆转或治愈严重或危及生命的疾病或病症;(3)初步临床证据表明,该药物有可能解决此类疾病或病症未满足的医疗需求。CBER对于获得RMAT认定的药物有相应的政策和审评程序,以提供加速研发和审评的组织保
六、政策特点及效果
美国干细胞疗法监管呈现“联邦严控vs州级放行”的双轨制。联邦层面纳入药品管理:美国将细胞治疗纳入药品法规,接受FDA监管。依据《21世纪治愈法案》设立再生医学先进疗法(RMAT)认定,针对严重疾病的再生医学产品,允许临床试验早期与FDA高频沟通优化研发。截至2025年6月,FDA共收到317项RMAT申请,151项获批。2024年12月,FDA批准同种异体骨髓间充质干细胞疗法Ryoncil用于治疗>2月龄儿童类固醇难治性急性移植物抗宿主病(SR-aGVHD),成为首款间充质干细胞药物,验证了RMAT路径可行性。FDA已出台20多个技术性指导原则直接指导干细胞产品研发。州级层面依托州立法开展尝试:以“患者自主权”为由立法扩大治疗选择,加速临床转化与商业化。该模式为资金不足的生物公司提供研发机会,同时让患者更早接触创新疗法,但也引发伦理安全争议,导致未充分验证疗法入市增加患者风险。
未来挑战:面对干细胞疗法的快速发展和州级立法的挑战,美国联邦政府通过加强监管、支持创新和打击非法行为等措施,不断调整干细胞监管策略,以适应快速发展的技术需求。北美干细胞产业仍是全球最大的市场,美国加州政府已投入30亿美元用于干细胞基础研究,带动约5.6万个就业岗位,创造经济效益超过100亿美元。2024年底,FDA批准了首款间充质干细胞药物,标志着美国干细胞疗法进入商业化关键阶段。整体来看,美国干细胞产业在政策支持、技术创新和市场需求的共同推动下,未来有望继续保持高速增长。
评论