

牙科|口腔等定制器械|出口欧盟CE市场准入流程指南详解|定制器械CE认证流程全解

定制器械出口欧洲--MDR CE
MDR定制器械(Custom-made Devices, CMDs)要点速览
一、核心定义(MDR 法规原文精简)
定制器械:指依据合法授权人员(医生/牙医等)的书面处方,利用其专业知识专门制造;具备独特设计特性,专供某一特定患者使用,以满足其个性化医疗需求的器械。
二、三大判定标准(缺一不可)
1. 专业处方:必须有授权医疗人员的书面指令。
2. 特定患者:产品唯一对应单个患者,非通用。
3. 非大规模生产:单件/小批量定制,非工业流水线。
三、易混淆概念(非定制器械)
以下两类虽为“量身定制”,但不属于 CMDs,需走常规上市路径:
1. 适应性器械:对量产器械进行调整(如调节轮椅、定制眼镜架)。
2. 患者匹配器械:基于患者数据(如3D扫描),但通过工业流程批量生产的标准化产品。
四、监管路径差异
- 定制器械(CMDs):豁免CE认证,需合规声明+技术文档,遵循简化监管。
- 适应性/患者匹配器械:按常规MDR流程,必须CE认证,走标准上市通道。
欧盟法规-定制器械申办路径和制造商义务-法规原文解析


总结:
定制器械出口欧盟需要完成CE认证路径归纳满足以下列出的所有要求,完成器械注册后方可上市销售。主要包括以下几个方面:
1、制定欧盟授权代表,签署欧盟授权代表协议
2、编写欧盟技术文件,建立符合 MDR 法规附录 X 第 2 节要求的技术文件。

3、履行制造商义务
a.建立实施风险管理体系
b.建立实施质量管理体系
c.临床评价
d.上市后监管体系(含警戒系统)
e.技术文件
f. 遵循附录 X 程序,无 CE 标识
9.纠正预防措施系统
h.根据欧盟成员国要求上交其境内上市的器械目录
4、完成Doc符合性说明
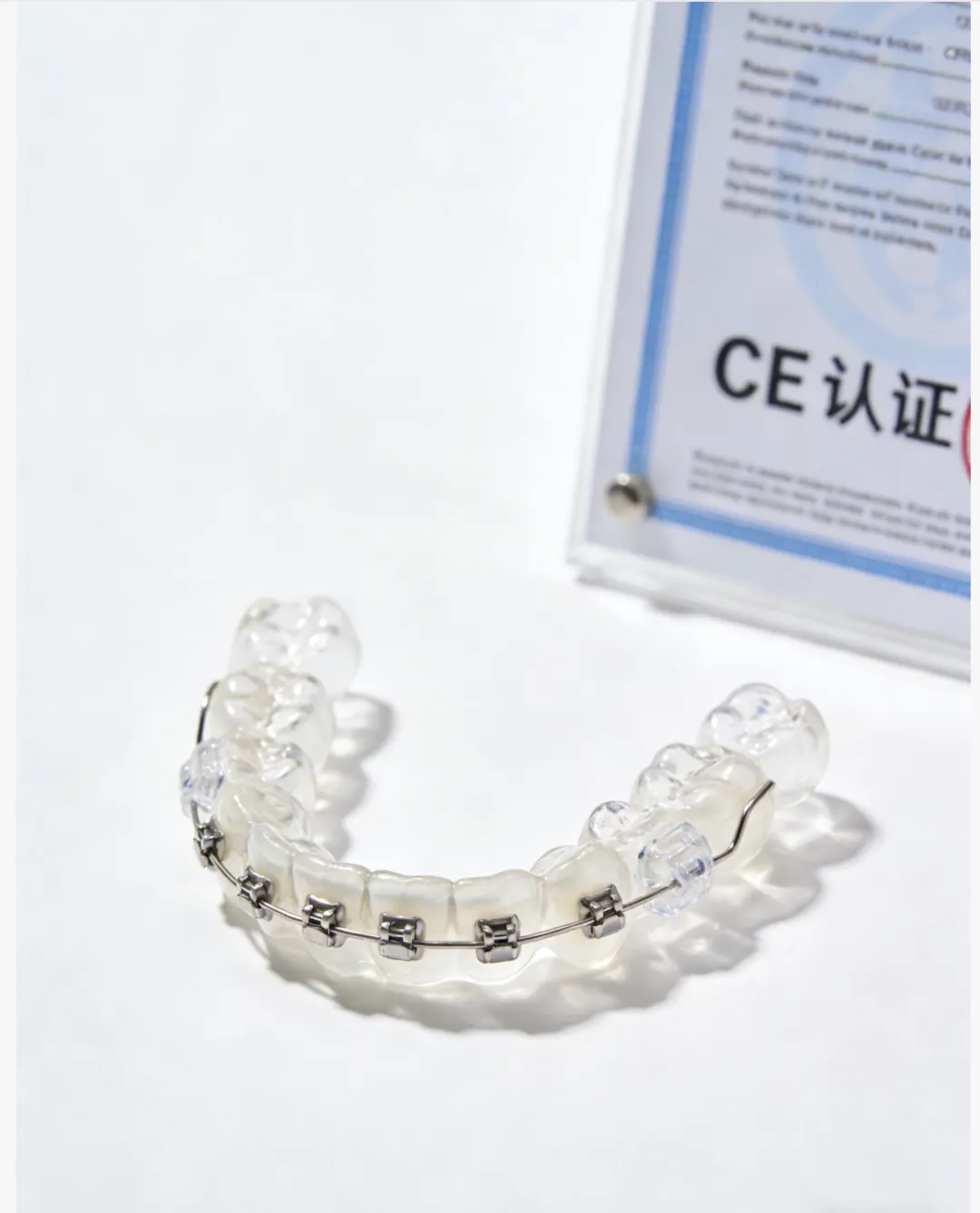
产品举例:隐形矫治器和保持器产品(口腔内持续逗留时间超过30天)
MDR定制器械出口欧盟,别让合规难题阻碍市场机遇,定制器械出口欧盟CE认证联系萬易集围(香港)醫療科技有限公司18372108953。
万易咨询官网:www.winning-medical-group.com

万易咨询Winning一站式服务:专业评估、文件编制、欧盟代表、全程跟进,省心高效、通过率高,助力中国定制器械合规通行欧盟。
定制器械出口欧盟的合规要点,今天就为大家梳理到这里。
下一篇,我们将继续聚焦定制器械出海赛道,为您深度解析英国UKCA与澳大利亚TGA的准入规则与实操要点,助您全面布局全球高端市场,敬请期待

评论